■再生医療が普及することに伴うことが想定されるさまざまな社会への影響
科学の不確実性に起因する、社会への長期的影響を踏まえ、再生医療をどのような規制の下に置くべきか?
【アジェンダ】
再生医療にはプラスの面として劇的な治療効果が期待される反面、マイナス面として、科学の不確実性に起因する、社会への諸々の長期的影響についての懸念もあります(現在は予測できない危険性がずっと後になってから判明するなど)。この懸念を受けて、あなたは再生医療をどのような規制の下に置くべきと考えますか。
なお現在でもすでに再生医療には、他の医療技術(医薬品や医療機器など)と同じように、品質・有効性・安全性を確保するために薬事法とその関連法令・通知による規制が設けられ、製造・販売には厚生労働大臣の承認が必要とされています(*)。
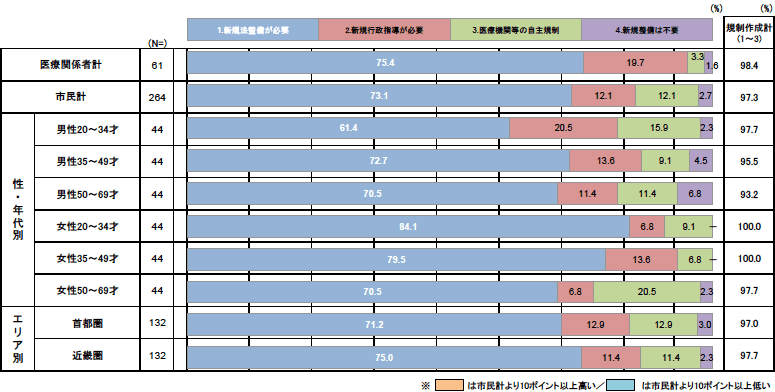
今後、再生医療をどのような規制の下に置くべきか、お答えください。
【ひとつ選択】
- 現在の規制に加えて、別途、再生医療を対象とした新しい法律による規制が必要(違反の場合は処罰も必要)。
- 現在の規制以外の法律化はしないが、行政が再生医療を対象とした指導を行うことが必要。
- 現在の規制に加え、医療機関や業界団体が自主規制を行う。
- 現在の規制を続けるだけでよい。
注(*)
現在の規制制度では、再生医療で用いる細胞・組織から加工した製品(例:培養皮膚、培養軟骨、培養角膜)、医薬品、医療機器は、他の一般の医薬品・医療機器等と同様に、薬事法とその関連法令・通知に基づいて、有効性と安全性に関する臨床試験(治験)を経て、その結果をもとに国の審査機関(独立行政法人医薬品医療機器総合機構)が審査し、厚生労働大臣の承認を経て、製造・販売が認可されることになっています。
また細胞・組織から加工した製品は、新規性が高いため過去の使用経験・情報の蓄積が少なく、リスクの予測が難しいことや、人間または動物由来の細胞・組織を用いることから、感染症の伝播の危険性が懸念されます。このため治験を行う前に「確認申請」という国の審査(独立行政法人医薬品医療機器総合機構が実施)を受け、品質と安全性を確保することが、「細胞・組織を利用した医療用具又は医薬品の品質及び安全性の確保について」(平成11年7月30日付け医薬発第906号、平成21年5月18日付け薬食発第0518001号にて改正)によって定められています。
さらに薬事法の規制がかからない「臨床研究」(研究者である医師が「医業」の範囲内で自主的に行うもの)の場合、通常は、安全性と倫理性を確保するために厚生労働省が定めた「臨床研究に関する指針」に従い、医療機関内に置かれた倫理審査委員会による審査を経て、機関の長が許可することで実施できますが、幹細胞を用いる場合には、「ヒト幹細胞を用いる臨床研究に関する指針」に従い、機関内の倫理審査に加えて、厚生労働大臣の了承が必要とされています。
«Q8
